News
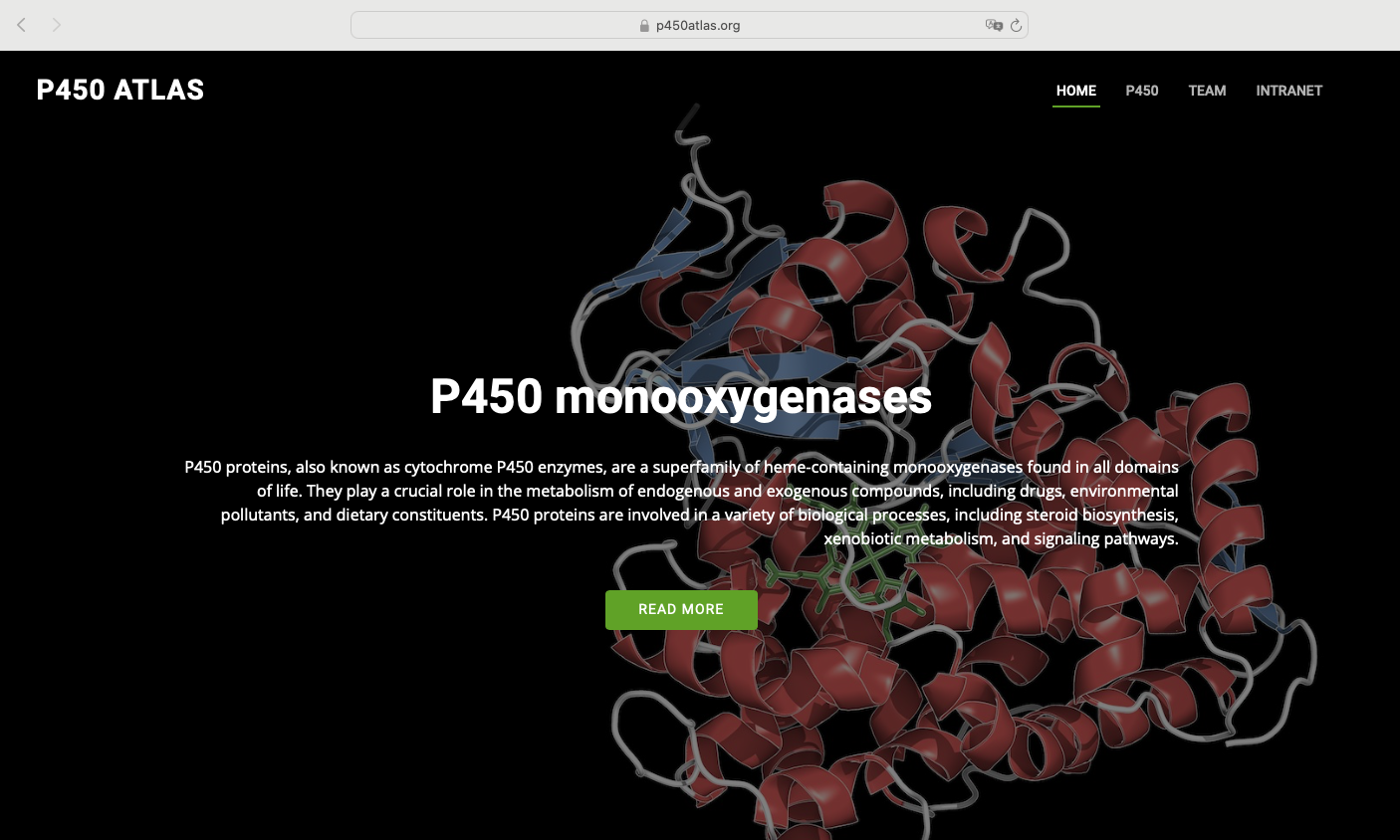
P450atlas website released!
The 16th International Symposium Cytochrome - P450 Biodiversity & Biotechnology featured the premiere of the P450atlas website - the ultimate reference for P450 enzyme
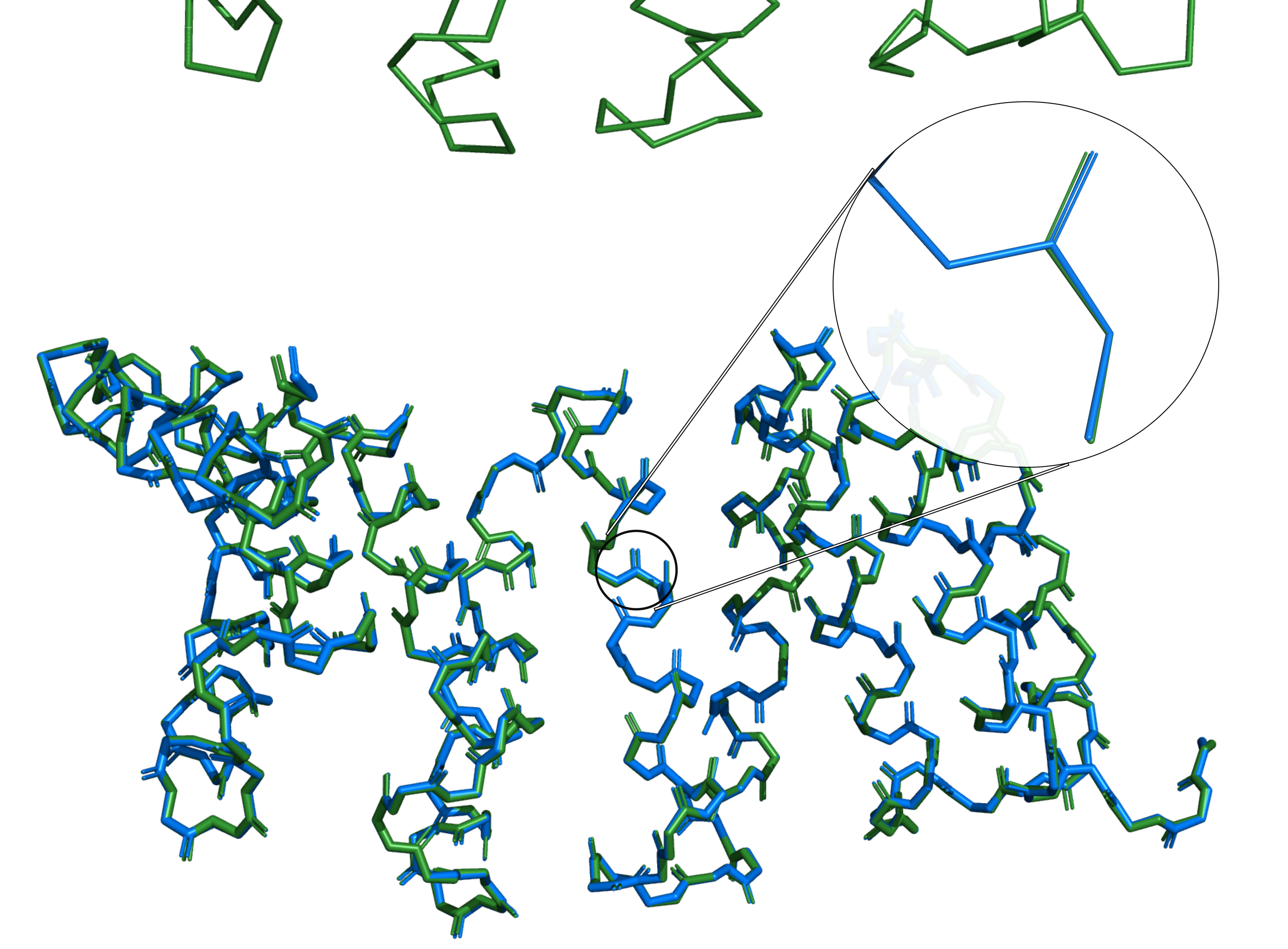
Our protein backbone reconstruction model deepBBQ published!
The article describing our new program for protein backbone reconstruction 'deepBBQ' has just been accepted for publication in 'Biomolecules' journal.

Exchange students from France
This summer two exchange students from France have visited our lab to work on the new version of the Bioshell package.
Projects
Projects developed in our group
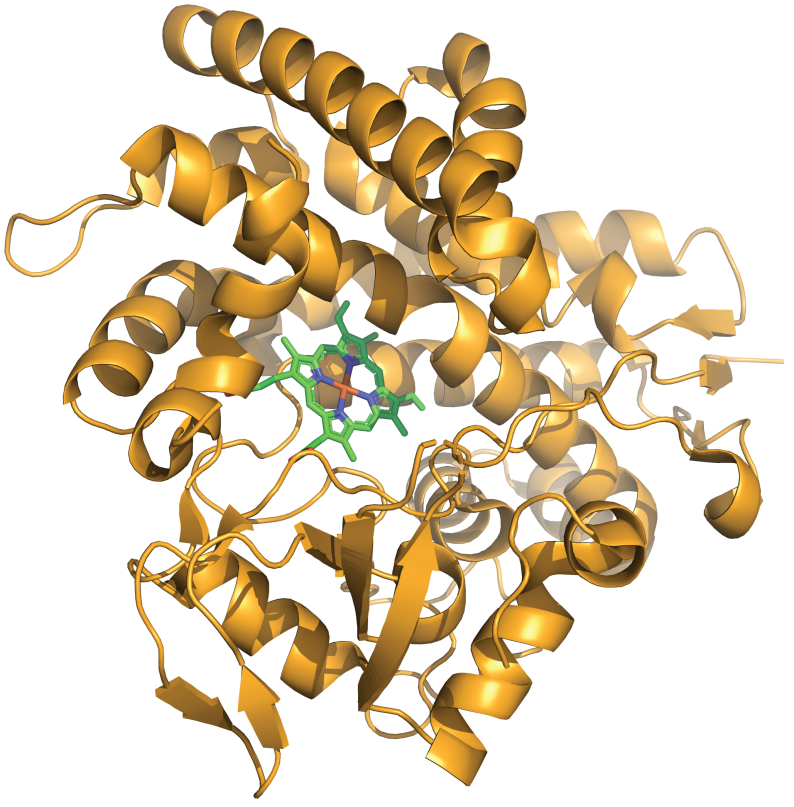
p450atlas
Cytochrome P450 enzymes are a superfamily of heme-containing monooxygenases found in all domains of life. They play a crucial role in the metabolism of endogenous and exogenous compounds, including drugs, environmental pollutants, and dietary constituents. P450Atlas provides an easy way to assign a family and subfamily for the user-provided P450 enzyme sequence.
Bioshell
BioShell is a general bioinformatics toolkit, focused on biomolecular structures. Its functionality covers file processing such as data filtering and file formats convertion. It handle protein sequences, sequence profiles and alignments. Structures calculations capabilities include superimpositions, crmsd calculations, alignments, Phi/Psi angles and many more.

SURPASS
Single United Residue per Pre-Averaged Secondary Structure fragment is a coarse-grained low resolution model for protein simulations. Its design has been based on the statistical analysis of structural regularities characteristic for protein systems. The SURPASS software is available free of charge to academic use on Apache 2.0 license.

Visualife
VisuaLife is a Python library for making interactive data visualization in a web-browser. It's focused on visualization of biological data such as sequence alignments and macromolecular structures, but can be used to create various kinds of plots and drawing.

Rosetta
Our lab, as a part of Rosetta Commons, actively participates in the development of Rosetta Macromolecular modelling toolkit. Rosetta software package has been a very successful method for protein design and modeling, docking,and many other applications!
Recent publications
All publicationsGront, D.;Syed, K.;Nelson, D.R. Exploring P450 superfamily diversity with P450Atlas - Online tool for automated subfamily assignment.. Protein science : a publication of the Protein Society. 2025, 34 (3), e70057. https://doi.org/10.1002/pro.70057
Kryś, J.D. et al. deepBBQ: A Deep Learning Approach to the Protein Backbone Reconstruction.. Biomolecules. 2024, 14 (11), 1448. https://doi.org/10.3390/biom14111448
Lab Members

Dominik Gront
Principal Investigator
Mohammad Saqib
PhD student
Piotr Śmieja
PhD studentCollaborators

Khajamohiddin Syed
University of Zululand
David Nelson
University of TennesseeContact
Email Us
dgront@chem.uw.edu.pl
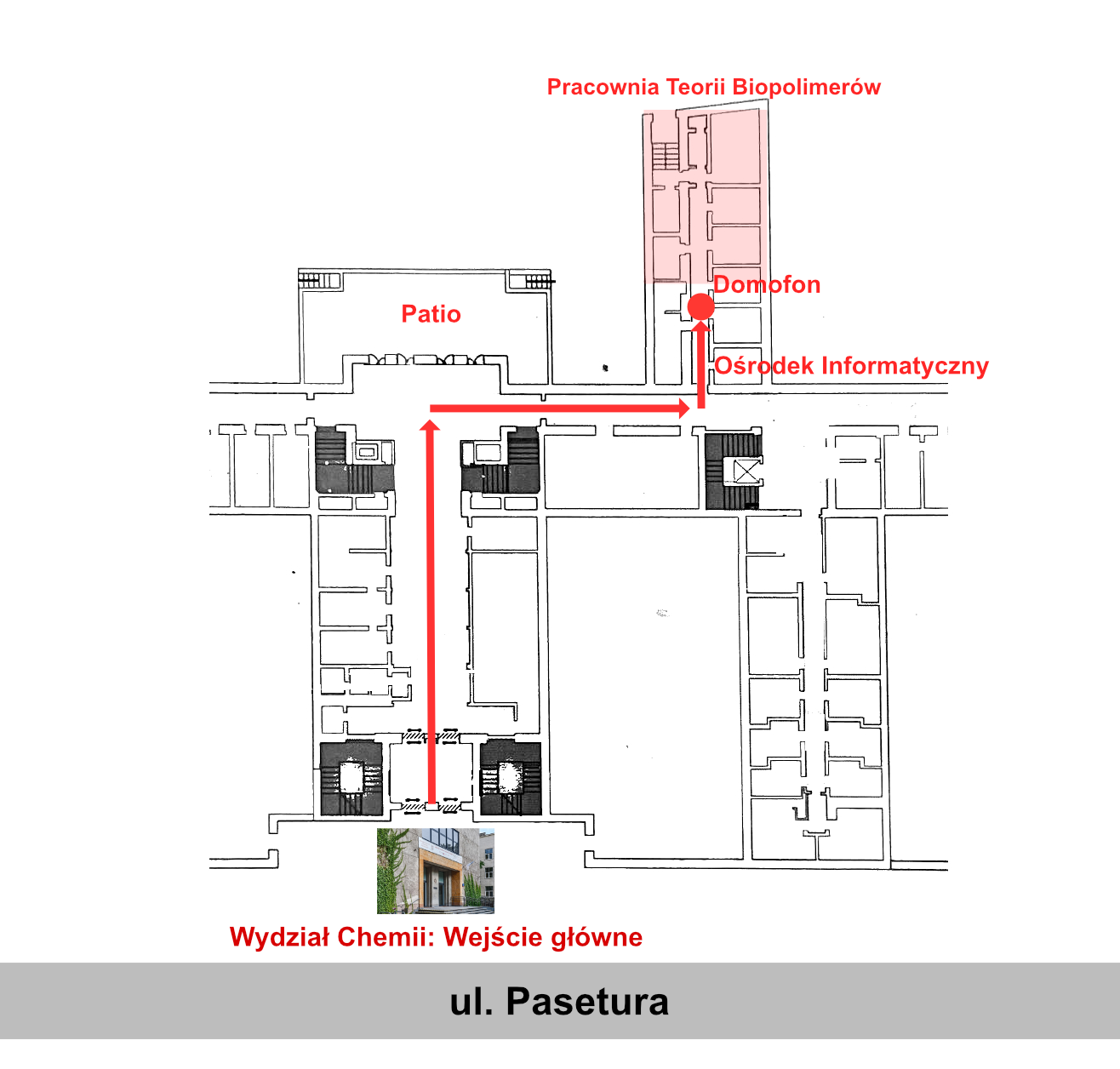
Address
Laboratorium Teorii Biopolimerów
Wydział Chemii Uniwersytetu Warszawskiego
ul. Pasteura 1, 02-093 Warszawa
Poland